
I
I
II
III
IV
II
III
IV








Durch eine Mutation ist die Erbinformation des GAL-Gens fehlerhaft und wird in ein defektes Protein übersetzt.
Das GAL-Gen im Zellkern liefert die Erbinformationen für die Bildung des Proteins Alpha-Galaktosidase A
Das Endoplasmatische Retikulum erkennt den Fehler und baut das Protein ab. Die Folge ist ein Mangel an Galaktosidase.
Durch das Fehlen des „Verdauungsenzyms“ können Sphingolipide nicht abgebaut werden und häufen sich im Lysosom an.
Dies führt zu Gewebeschäden in unterschiedlichsten Organsystemen und verursacht das komplexe Krankheitsbild bei Morbus Fabry.
Therapie:
Patienten mit Morbus Fabry wird das fehlende Enzym Alpha-Galaktosidase A dem Körper regelmäßig per Infusion verabreicht.
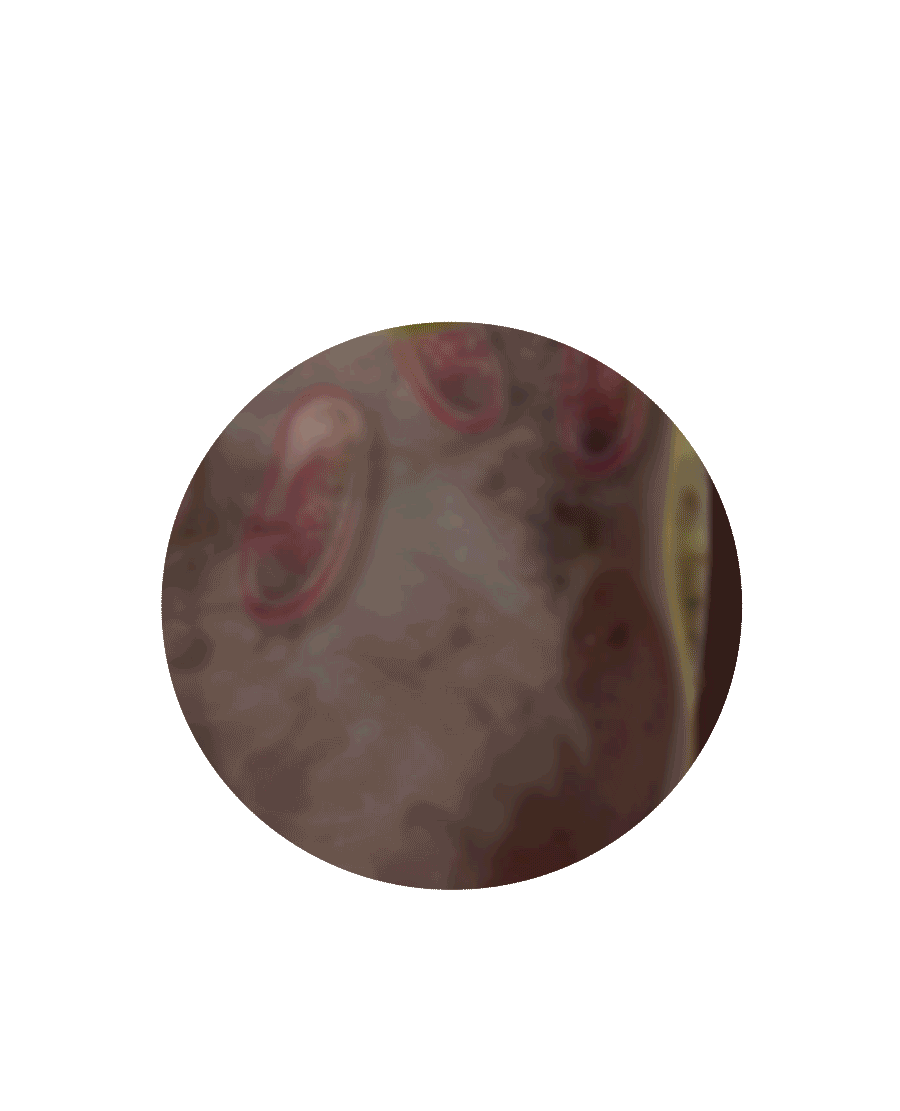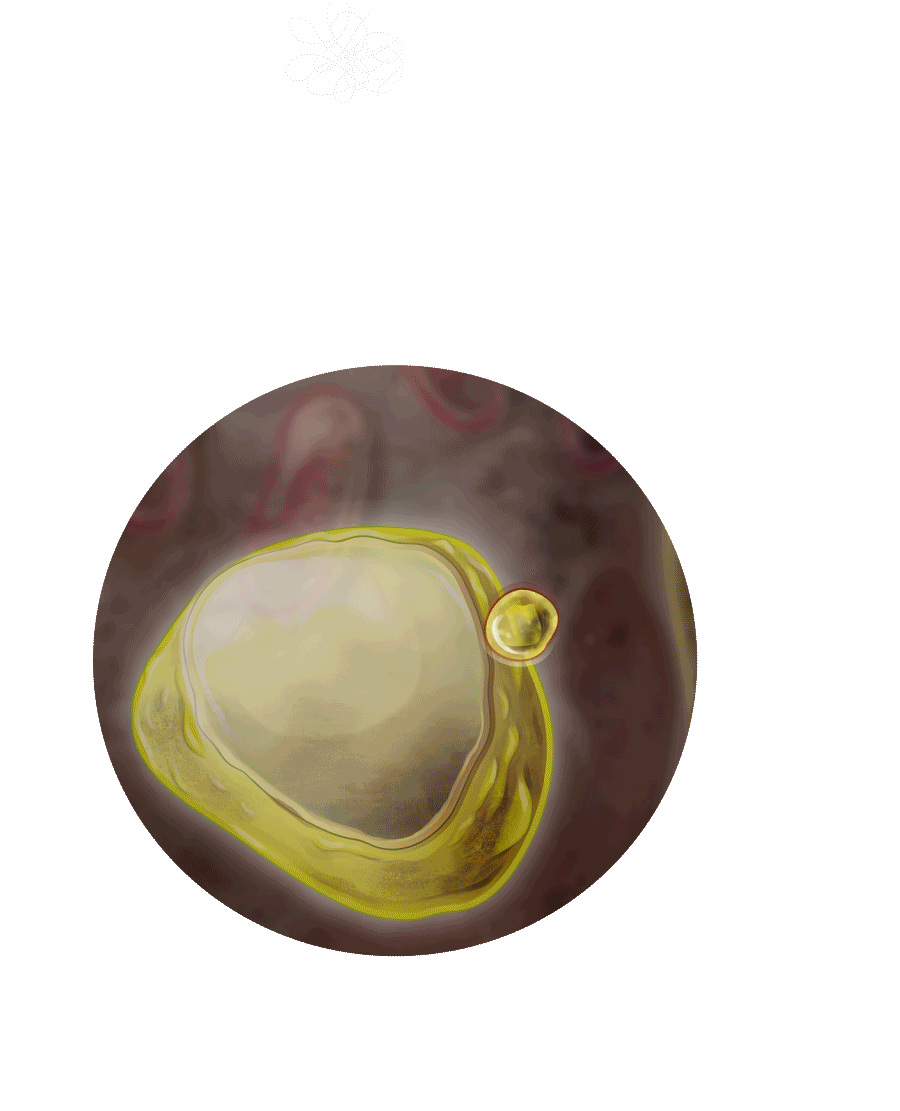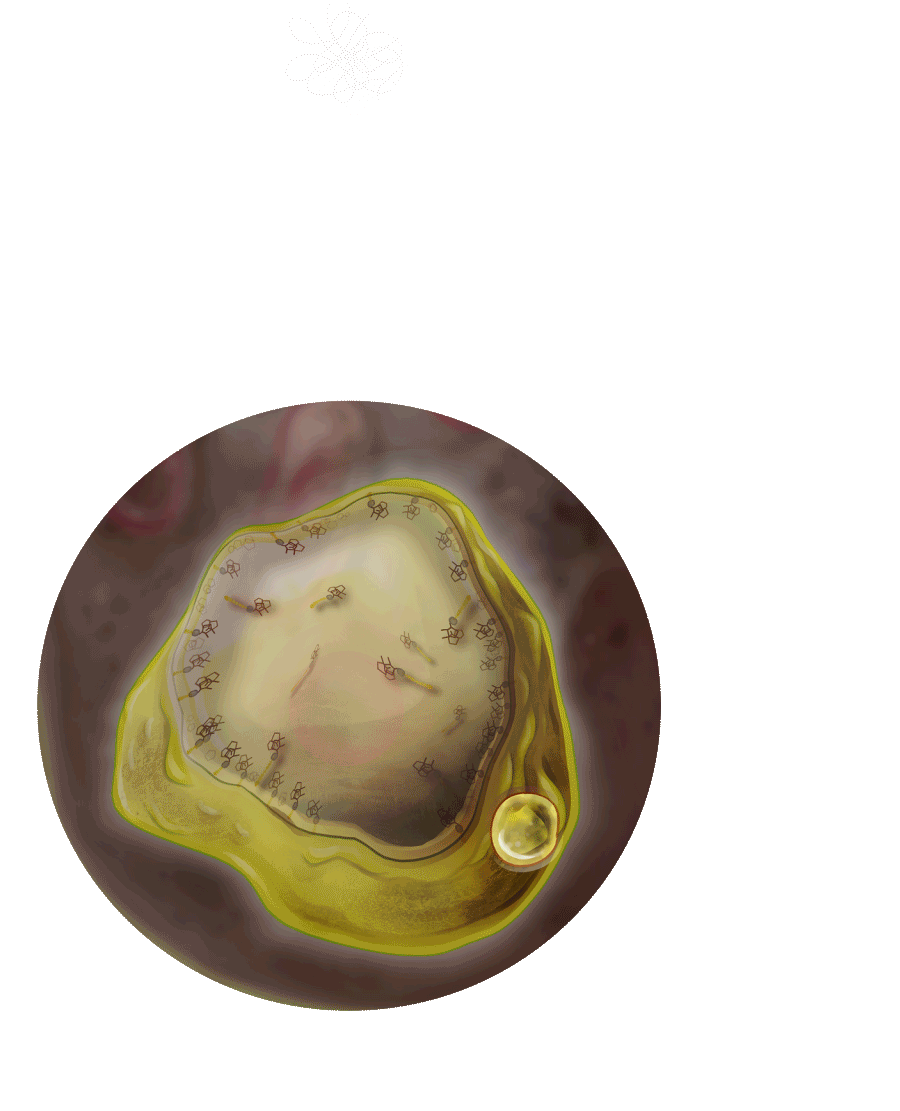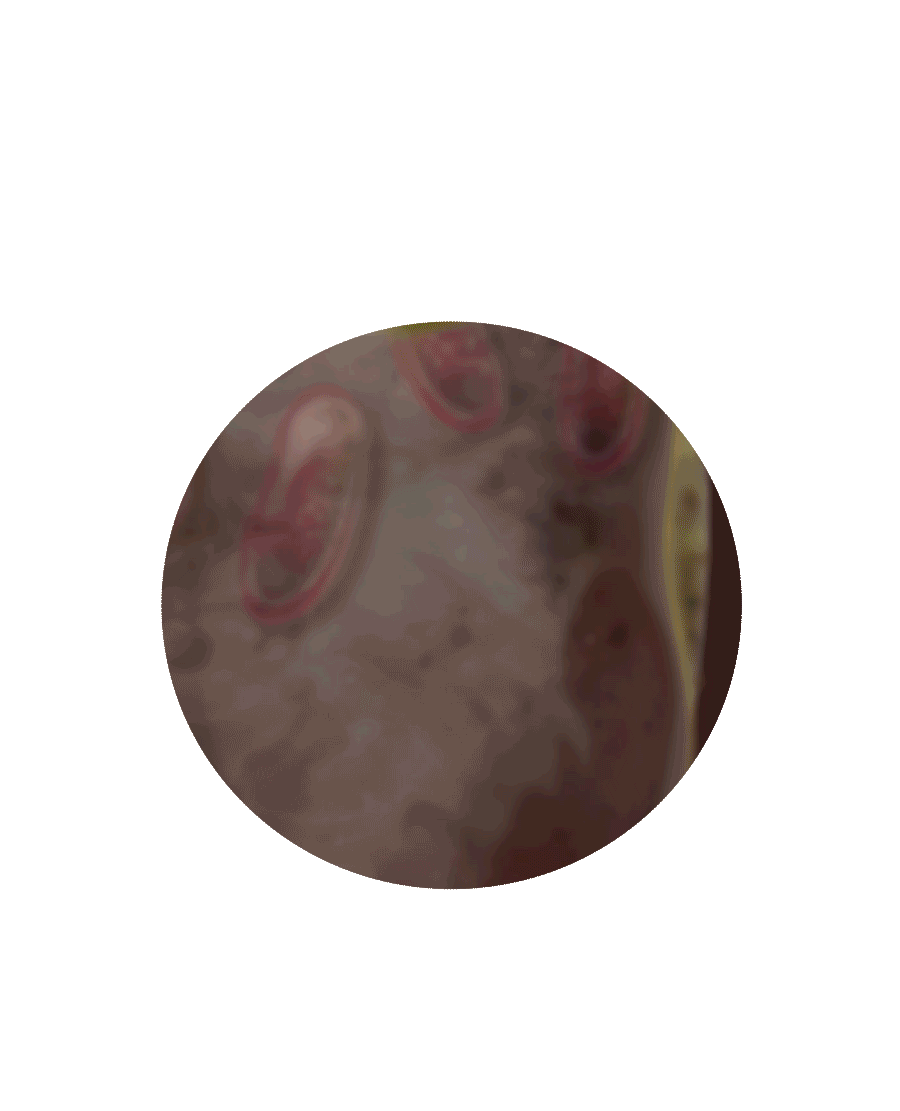
Nach bestandener Prüfung gelangt die Galaktosidase ins Lysosom.
Auch Bestandteile der äußeren Zellmembran, die sogenannten Sphingolipide,
wandern ins Lysosom.
Als „Verdauungsenzym“ hilft die Galaktosidase beim Abbau der Sphingolipide.
Das Protein durchläuft eine Prüfung im Endoplasmatischen Retikulum
Morbus Fabry – das geschieht im Inneren der Zelle
Morbus Fabry gehört zu den Lysosomalen Speicherkrankheiten.
Diesen Stoffwechselerkrankungen liegt eine Störung im Lysosom zugrunde, dem „Magen der Zelle“.
Bei Patienten mit Morbus Fabry, fehlt dem Lysosom das Enzym Alpha-Galaktosidase A.
Die Folge: Stoffwechselprodukte können nicht richtig abgebaut werden und lagern sich in den Zellen ab.
Das führt zu Symptomen an unterschiedlichen Organsystemen wie Nieren, Augen, Herz und Haut.
Bewegen Sie die Maus der Reihe nach über die nummerierten Kreise
und erfahren Sie Schritt für Schritt, welche zellulären Mechanismen bei Mobus Fabry verändert sind.
Gesundes Gen
Morbus Fabry
Zellkern
Lysosom
Mitochondrium
Endoplasmatisches
Retikulum